/ Amsterdam UMC / NSCLC /
Early stage SBRT-immuno
Combining SBRT and immunotherapy in early stage NSCLC patients planned for surgery: exploring safety and immunological proof of principle.
Population
Patients with early stage (T1N0 and T2aN0) peripherally located NSCLC, eligible for surgical resection.
Design
An open label, 1:1:1 randomized, exploratory study
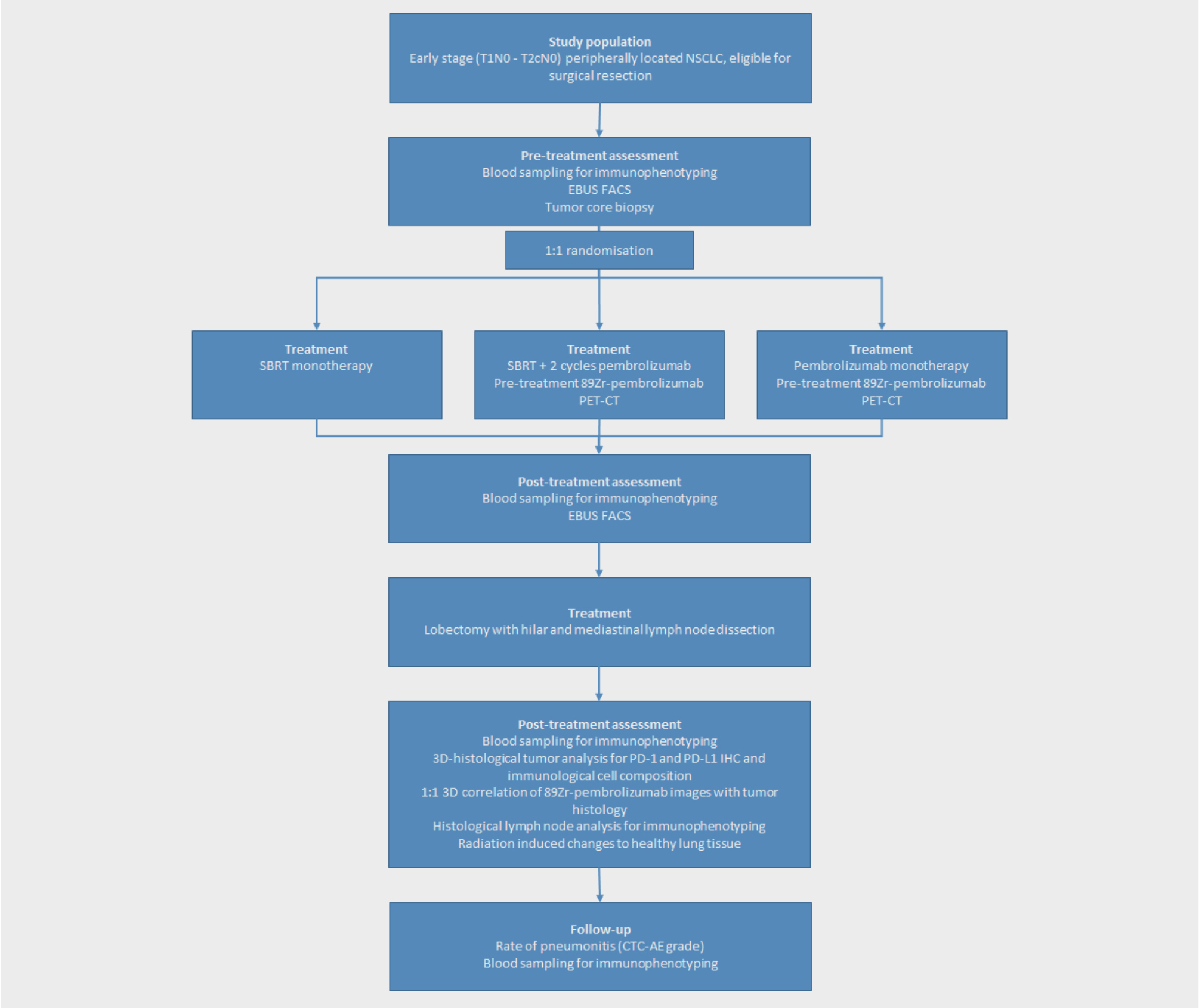
Key Outcome parameters
Primary endpoints:
- Safety: Incidence and severity of adverse events in patients treated by combining SBRT and pembrolizumab
- Mechanism of action: To identify the immunological response to combined SBRT and pembrolizumab treatment in early stage NSCLC and compare this to the immunological response to SBRT and pembrolizumab monotherapy
Intervention
Arm A: SBRT prior to surgery,
Arm B: SBRT with 2 cycles of pembrolizumab treatment (starting on the first day of radiotherapy) prior to surgery
Arm C: 2 cycles of pembrolizumab monotherapy as induction regimen prior to surgery.
Patients will undergo a lobectomy with hilar and mediastinal lymph node dissection after induction therapy. Translational research to explore the immune mechanism of action will include biological imaging with immuno-PET. Expression rates and activation states of immune effector subsets will be assessed in tumor core biopsy specimens, peripheral blood and tumor draining lymph nodes (TDLNs) by means of fine needle aspirates of TDLNs. Samples will be taken before and after induction therapy and at surgery.
Key inclusion criteria
- A histologically or cytologically confirmed diagnosis of early stage (T1bN0 and T2aN0) peripherally located NCSLC, eligible for surgical resection
- Have measurable disease based on RECIST 1.1.
- Must provide tissue from a core or excisional biopsy of the primary tumor lesion.
Key exclusion criteria
- currently participating in or has participated in a study of an investigational agent or using an investigational device within 4 weeks of the first dose of treatment.
- Has a diagnosis of immunodeficiency or is receiving systemic steroid therapy or any other form of immunosuppressive therapy within 7 days prior to the first dose of trial treatment.
- Has had a prior monoclonal antibody within 4 weeks prior to study Day 1 or who has not recovered (i.e., ≤ Grade 1or at baseline) from adverse events due to agents administered more than 4 weeks earlier.
- Has had prior chemotherapy, targeted small molecule therapy, or radiation therapy within 2 weeks prior to study Day 1 or who has not recovered (i.e., ≤ Grade 1 or at baseline) from adverse events due to a previously administered agent.
- Note: Subjects with ≤ Grade 2 neuropathy are an exception to this criterion and may qualify for the study.
- Note: If subject received major surgery, they must have recovered adequately from the toxicity and/or complications from the intervention prior to starting therapy.
- Has a known additional malignancy that is progressing or requires active treatment. Exceptions include basal cell carcinoma of the skin, squamous cell carcinoma of the skin, or in situ cervical cancer that has undergone potentially curative therapy.
- Has an active autoimmune disease requiring systemic treatment within the past 3 months or a documented history of clinically severe autoimmune disease, or a syndrome that requires systemic steroids or immunosuppressive agents. Subjects with vitiligo or
resolved childhood asthma/atopy would be an exception to this rule. Subjects that require intermittent use of bronchodilators or local steroid injections would not be excluded from the study. Subjects with hypothyroidism stable on hormone replacement or Sjorgen’s
syndrome will not be excluded from the study. - Has a history of (non-infectious) pneumonitis that required steroids, evidence of interstitial lung disease or active, non-infectious pneumonitis.
- Has an active infection requiring systemic therapy. 9. Has a history or current evidence of any condition, therapy, or laboratory abnormality that might confound the results of the trial, interfere with the subject’s participation for the full duration of the trial, or is not in the
best interest of the subject to participate, in the opinion of the treating investigator. - Has known psychiatric or substance abuse disorders that would interfere with cooperation with the requirements of the trial.
- Has received prior therapy with an anti-PD-1, anti-PD-L1, anti-PD-L2, anti-CD137, or anti-Cytotoxic T-lymphocyte- associated antigen-4 (CTLA-4) antibody (including ipilimumab or any other antibody or drug specifically targeting T- cell co-stimulation or checkpoint pathways).
- Has a known history of Human Immunodeficiency Virus (HIV) (HIV 1/2 antibodies).
- Has known active Hepatitis B (e.g., HBsAg reactive) or Hepatitis C (e.g., HCV RNA [qualitative] is detected).
- Has received a live vaccine within 30 days prior to the first dose of trial treatment.
Contact opnemen over een studie
Neem contact op voor meer informatie over de studies van de afdeling thoracale oncologie van Amsterdam UMC.