PACIFIC-4
A Phase III, Randomized, Placebo-controlled, Double-blind, Multi-center, International Study of Durvalumab Following Stereotactic Body Radiation Therapy (SBRT) for the Treatment of Patients with unresected Stage I/II, lymph-node negative Non-small Cell Lung Cancer (PACIFIC-4/RTOG-3515)
Enrollment
Recruiting, both cohorts will probably close enrolment in June 2024
No. of patients
1 / 630
Population
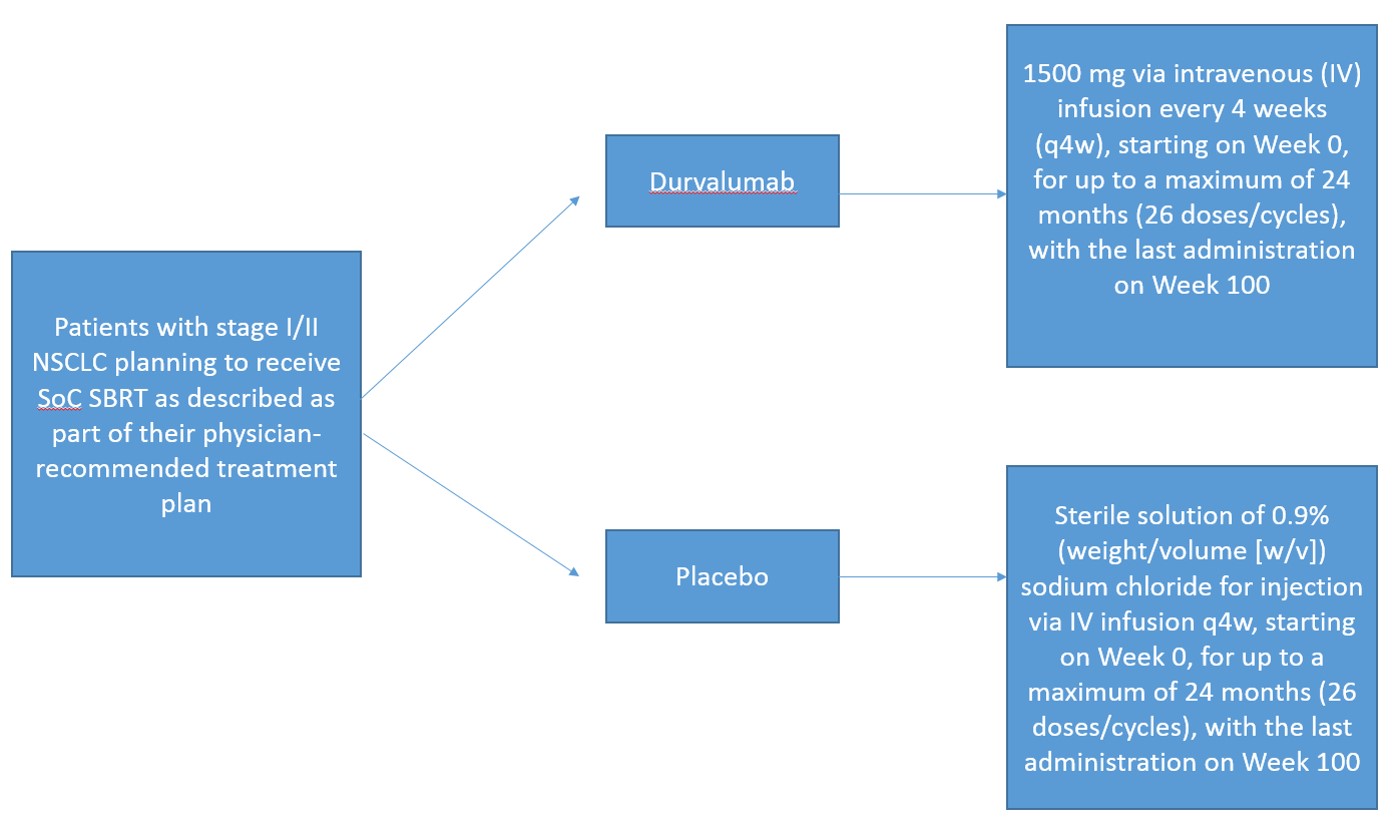
In order to be eligible for this study, patients must be planning to receive SoC SBRT as described as part of their physician-recommended treatment plan. Prior to SoC SBRT treatment, patients will provide a baseline blood sample for circulating tumor DNA (ctDNA) assessment and undergo a baseline computed tomography (CT) scan. These patients will then undergo definitive treatment with SoC SBRT as planned
Design
This is a Phase III, randomized, placebo-controlled, double-blind, multi-center study assessing the efficacy and safety of durvalumab versus placebo following SoC SBRT in patients with unresected clinical Stage I/II lymph node-negative (T1 to T3N0M0) NSCLC
Key outcome parameters
Primary objective:
- To assess the efficacy of durvalumab monotherapy compared to placebo in terms of PFS in patients with T1c to T3N0M0 NSCLC
Endpoint/variable:
- PFS using BICR assessments according to RECIST 1.1
Secondary objectives:
- To assess the efficacy of durvalumab monotherapy compared to placebo in terms of PFS in patients with Stage I/II NSCLC
- To assess the efficacy of durvalumab monotherapy compared to placebo in terms of OS
- To further assess the efficacy of durvalumab monotherapy compared to placebo in terms of lung cancer-specific mortality
- To assess the PK of durvalumab
- To investigate the immunogenicity of durvalumab
- To assess symptoms and health-related quality of life in patients treated with durvalumab monotherapy compared to placebo using the EORTC QLQ-C30
Endpoints/variables:
- PFS using BICR assessments according to RECIST 1.1
- OS in patients with T1c to T3N0M0 NSCLC
- OS in patients with Stage I/II NSCLC
- Lung cancer mortality
- PFS24a, TTP, and TTDM using BICR assessments according to RECIST 1.1
- PFS2 using local assessment
- Concentration of durvalumab in blood such as peak concentration and trough, as data allow(sparse sampling)
- Presence of ADA for durvalumab (confirmatory results: Positive or negative; titers)
- EORTC QLQ-C30: Change in symptoms, functioning, and global health status/quality of life
Intervention
- Durvalumab 1500 mg via intravenous (IV) infusion every 4 weeks (q4w), starting on Week 0, for up to a maximum of 24 months (26 doses/cycles), with the last administration on Week 100
- Placebo: Sterile solution of 0.9% (weight/volume [w/v]) sodium chloride for injection via IV infusion q4w, starting on Week 0, for up to a maximum of 24 months (26 doses/cycles), with the last administration on Week 100
Key inclusion criteria
Capable
- of giving signed informed consent, which includes compliance with the requirements and restrictions listed in the informed consent form (ICF) and in this
protocol - Provision of signed and dated written ICF prior to any mandatory study specific procedures, sampling, and analyses
- Provision of signed and dated written genetic informed consent prior to collection of sample for genetic analysis
- Age ≥18 years at the time of screening. For patients aged <20 years and enrolled in Japan, a written informed consent should be obtained from the patient and his or her
legally acceptable representative - Histologically or cytologically documented Stage I to II NSCLC, with clinical Stage I/II lymph node-negative (T1 to T3N0M0) disease and planned to receive definitive treatment with SBRT. In order to be eligible for this trial, patients should be:
- Medically inoperable as determined by physician
- Medically operable with patient refusal of surgery
- Patients with medically operable disease who choose to have SBRT are also eligible
- Completion of SoC SBRT as definitive treatment prior to randomization using one of the following doses:
- For peripheral tumors: 54 Gy total dose delivered in 3 fractions, 42 Gy total dose delivered in 4 fractions, or 50 Gy total dose delivered in 5 fractions
- For central tumors: 50 Gy total dose delivered in 5 fractions
- World Health Organization (WHO)/Eastern Cooperative Oncology Group (ECOG) PS of 0, 1, or 2 at enrollment and randomization
- Tumor sample requirements:
- Mandatory provision of an archived tumor tissue block ≤6 months old (or a minimum of 6 newly cut unstained slides; if available, up to 20 newly cut slides are encouraged) (refer to the Laboratory Manual for details). If archival tumor block or slides are both unavailable, cell blocks from fine needle aspirate (FNA) samples will be accepted
- A recent (≤3 months) tumor biopsy is encouraged, provided that a biopsy procedure is technically feasible, and the procedure is not associated with unacceptable clinical risk
- For China: Only tumor slides should be provided
- Patients with central or peripheral lesions are eligible. Central lesions are defined as tumor within or touching the zone of the proximal bronchial tree, defined as a volume of 2 cm in all directions around the proximal bronchial tree (carina, right and left main bronchi, right and left upper lobe bronchi, intermedius bronchus, right middle lobe bronchus, lingular bronchus right, and left lower lobe bronchi). Patients with Ultra-central tumors are NOT eligible. Ultra-central tumors are defined as tumors abutting the trachea, mainstem bronchus, or esophagus.
- Adequate organ and marrow function as defined below prior to randomization:
- Hemoglobin ≥9.0 g/dL
- Absolute neutrophil count ≥1.0 × 109/L
- Platelet count ≥75 × 109/L
- Serum bilirubin ≤1.5 × the upper limit of normal (ULN). This will not apply to patients with confirmed Gilbert’s syndrome, who will be allowed in consultation with their physician
- ALT and AST ≤2.5 × ULN within 2 weeks prior to randomization
- Measured creatinine clearance (CL) >30 mL/min or calculated CL >30 mL/min as determined by Cockcroft-Gault (using actual body weight)
Males: Creatinine CL = Weight (kg) × (140 – Age) (mL/min) 72 × serum creatinine (mg/dL)
Females: Creatinine CL = Weight (kg) × (140 – Age) × 0.85 (mL/min) 72 × serum creatinine (mg/dL)
- Must have a life expectancy of at least 12 weeks
- The following staging studies must be done within 8 weeks before randomization:
- Whole body positron emission tomography (PET)/CT scan from the top of the skull to mid-thigh is required for staging purposes. If PET scan not previously performed, may be added to baseline CT scan during screening to ensure adequate clinical staging
- Patients with hilar or mediastinal lymph nodes ≤1 cm and no abnormal hilar or mediastinal uptake on PET will be considered N0. Mediastinal lymph node sampling by any technique is allowed but not required. Patients with >1 cm hilar or mediastinal lymph nodes on CT or abnormal PET (including suspicious but nondiagnostic uptake) may still be eligible if directed tissue biopsies of all abnormally identified areas are negative for cancer
- Body weight >30 kg
- Male and/or female
Key exclusion criteria
- Mixed small cell and non-small cell cancer histology
- History of allogeneic organ transplantation
- Active or prior documented autoimmune or inflammatory disorders (including inflammatory bowel disease [eg, colitis or Crohn’s disease], diverticulitis [with the exception of diverticulosis], systemic lupus erythematosus, Sarcoidosis syndrome, or Wegener syndrome [granulomatosis with polyangiitis, Graves’ disease, rheumatoid arthritis, hypophysitis, uveitis, etc]). The following are exceptions to this criterion:
- Patients with vitiligo or alopecia
- Patients with hypothyroidism (eg, following Hashimoto syndrome) stable on hormone replacement
- Any chronic skin condition that does not require systemic therapy
- Patients without active disease in the last 5 years may be included but only after consultation with the Study Physician
- Patients with celiac disease controlled by diet alone
- Uncontrolled intercurrent illness, including but not limited to, ongoing or active infection, symptomatic congestive heart failure (NYHA Class III or IV), uncontrolled hypertension, unstable angina pectoris, uncontrolled cardiac arrhythmia, active interstitial lung disease, serious chronic gastrointestinal conditions associated with diarrhea, or psychiatric illness/social situations that would limit compliance with study requirement, substantially increase risk of incurring AEs, or compromise the ability of the patient to give written informed consent
- History of another primary malignancy except for:
- Malignancy treated with curative intent and with no known active disease ≥5 years before the first dose of IP and of low potential risk for recurrence
- Adequately treated non-melanoma skin cancer or lentigo maligna without evidence of disease
- Adequately treated carcinoma in situ without evidence of disease
- Patients with a history of metachronus NSCLC treated only with curative surgery >1 year prior to randomization are eligible. Patients with a previous history of NSCLC treated with chemotherapy and/or radiotherapy are not eligible
- History of active primary immunodeficiency
- Active infection including tuberculosis (clinical evaluation that includes clinical history, physical examination and radiographic findings, and tuberculosis testing in line with local practice), hepatitis B (known positive hepatitis B surface antigen [HBsAg] result), hepatitis C, or human immunodeficiency virus (positive human immunodeficiency virus [HIV] 1/2 antibodies). Patients with a past or resolved hepatitis B (HBV) infection (defined as the presence of hepatitis B core antibody [anti-HBc] and absence of HBsAg) are eligible. Patients positive for hepatitis C (HCV) antibody are eligible only if polymerase chain reaction is negative for HCV RNA
- History of non-infectious pneumonitis requiring steroids, or patients with Grade ≥2 pneumonitis
- Any unresolved toxicity National Cancer Institute (NCI) CTCAE Grade ≥2 from SBRT, with the exception of the laboratory values defined in the inclusion criteria:
- Patients with irreversible toxicity not reasonably expected to be exacerbated by treatment with IP may be included only after consultation with the Study Physician
- Known allergy or hypersensitivity to any of the study drugs or any of the study drug excipients Prior exposure to immune-mediated therapy including, but not limited to, other anti-CTLA-4, anti-PD-1, anti-PD-L1, and anti-programmed cell death ligand 2 antibodies, excluding therapeutic anticancer vaccines
- Any concurrent chemotherapy, IP, biologic, or hormonal therapy for cancer treatment. Concurrent use of hormonal therapy for non-cancer-related conditions (eg, hormone replacement therapy) is acceptable
- Receipt of live attenuated vaccine within 30 days prior to the first dose of IP. Note: Patients, if enrolled, should not receive live vaccine while receiving IP and up to 30 days after the last dose of IP
- Major surgical procedure (as defined by the Investigator) within 28 days prior to the first dose of IP
- Current or prior use of immunosuppressive medication within 14 days before the first dose of IP. The following are exceptions to this criterion:
- Intranasal, inhaled, topical steroids, or local steroid injections (eg, intra-articular injection)
- Systemic corticosteroids at physiologic doses not to exceed 10 mg/day of prednisone or its equivalent
- Steroids as premedication for hypersensitivity reactions (eg, CT scan premedication)
- Participation in another clinical study with an IP administered in the last 4 weeks
- Previous IP assignment in the present study
- Concurrent enrollment in another clinical study, unless it is an observational (non-interventional) clinical study or during the follow-up period of an interventional study
- Prior randomization or treatment in a previous durvalumab clinical study regardless of treatment group assignment
- Involvement in the planning and/or conduct of the study (applies to both AstraZeneca staff and/or staff at the study site)
- Female patients who are pregnant or breastfeeding or male or female patients of reproductive potential who are not willing to employ effective birth control from screening to 90 days after the last dose of durvalumab monotherapy
- Judgment by the Investigator that the patient should not participate in the study if the patient is unlikely to comply with study procedures, restrictions, and requirements.
- Exclusion criteria for participation in the optional (DNA) genetics research component of the study include:
- Previous allogeneic bone marrow transplant
- Non-leukocyte-depleted whole blood transfusion in 120 days of genetic sample collection
Contact opnemen over een studie
Neem contact op voor meer informatie over de studies van de afdeling thoracale oncologie van Amsterdam UMC.