ADRIATIC
A Phase III, Randomized, Double-blind, Placebo-controlled, Multi-center, International Study of Durvalumab or Durvalumab and Tremelimumab as Consolidation Treatment for Patients with Stage I-III Limited Disease Small-Cell Lung Cancer Who Have Not Progressed Following Concurrent Chemoradiation Therapy (ADRIATIC)
Enrollment
Not recruiting
No. of patients
600 / 724
Population
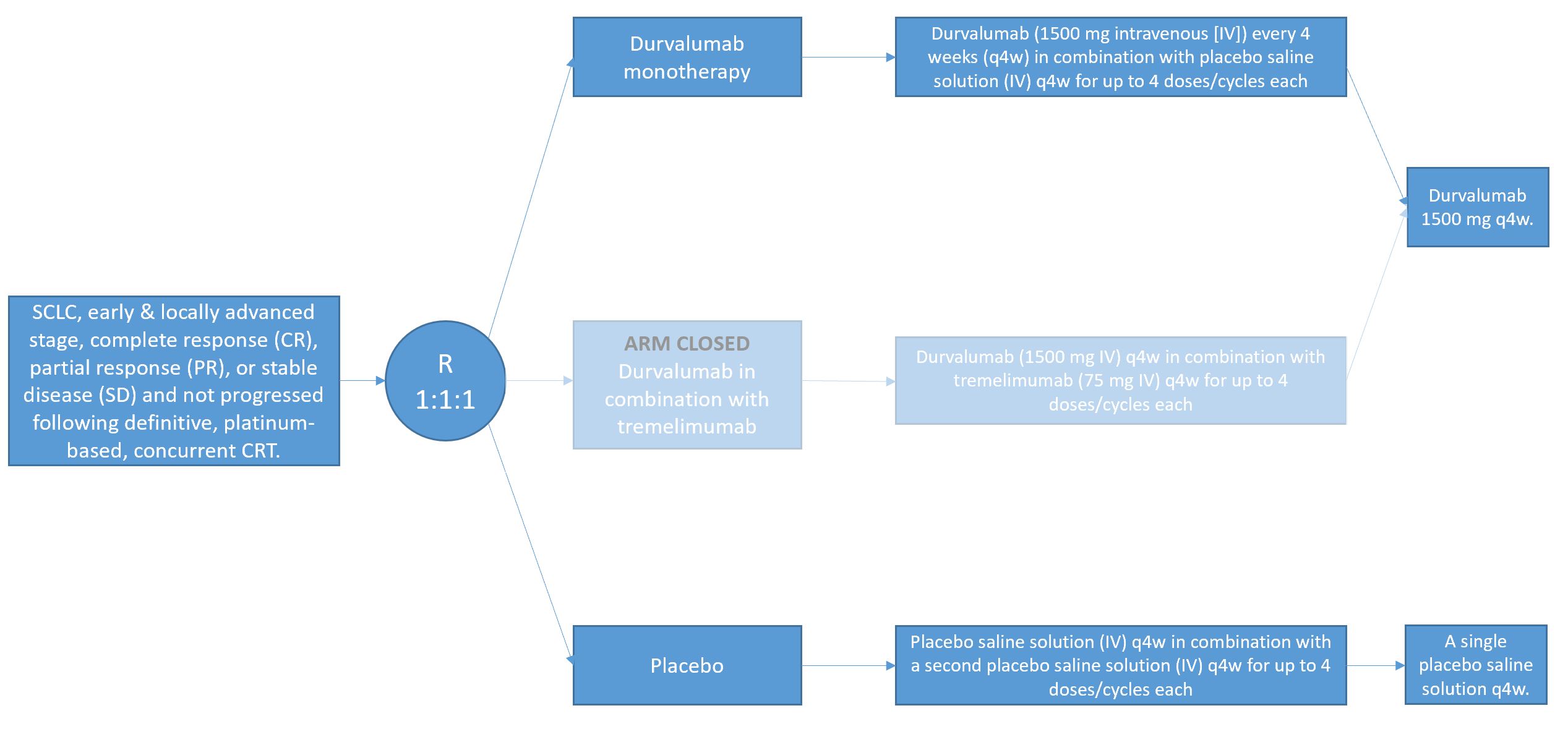
Patients who achieved complete response (CR), partial response (PR), or stable disease (SD) and have not progressed following definitive, platinum-based, concurrent CRT.
Design
A Phase III, 1:1:1 randomized, double-blind, placebo-controlled, multi-center study
Key Outcome parameters
Primary endpoints:
- PFS using BICR assessments according to RECIST 1.1
- OS
Secondary Objectives:
- To assess the efficacy of durvalumab monotherapy compared to placebo in terms of OS
- To further assess the efficacy of durvalumab monotherapy and durvalumab and tremelimumab combination therapy compared to placebo in terms of ORR, PFS18a , PFS24a , TTDM, OS24, OS36, and PFS2
- To assess the efficacy of durvalumab and tremelimumab combination therapy compared to durvalumab monotherapy in terms of PFS, OS, and ORR
- To assess disease-related symptoms and HRQoL in patients treated with durvalumab monotherapy or durvalumab and tremelimumab combination therapy compared to placebo using the EORTC QLQ-C30 v3 and QLQ-LC13
- To assess the PK of durvalumab monotherapy and durvalumab and tremelimumab combination therapy
- To investigate the immunogenicity of durvalumab monotherapy and durvalumab and tremelimumab combination therapy
- To investigate the relationship between a patient’s tumor mutational burden (TMB) measured in tumor and/or blood and efficacy outcomes with durvalumab and durvalumab and tremelimumab combination therapy
Intervention
- Durvalumab monotherapy: Durvalumab (1500 mg intravenous [IV]) every 4 weeks (q4w) in combination with placebo saline solution (IV) q4w for up to 4 doses/cycles each, followed by durvalumab 1500 mg q4w. The first durvalumab monotherapy 1500 mg dose q4w will be 4 weeks after the final dose of durvalumab in combination with placebo saline solution.
- Durvalumab in combination with tremelimumab: Durvalumab (1500 mg IV) q4w in combination with tremelimumab (75 mg IV) q4w for up to 4 doses/cycles each, followed by durvalumab 1500 mg q4w. The first durvalumab monotherapy 1500 mg dose q4w will be 4 weeks after the final dose of durvalumab in combination with tremelimumab.
- Placebo: Placebo saline solution (IV) q4w in combination with a second placebo saline solution (IV) q4w for up to 4 doses/cycles each, followed by a single placebo saline solution q4w. The first placebo saline solution monotherapy dose q4w will be 4 weeks after the final dose of the 2 placebo saline solutions in combination.
Key inclusion criteria
- Histologically or cytologically documented limited-stage SCLC (Stage I-III SCLC[T any, N any, M0], ie patients whose disease can be encompassed within a radical radiation portal. Patients who are Stage I or II must be medically inoperable.
- Performance status (PS) of 0 or 1 at enrollment and randomization
- Received 4 cycles of platinum-based chemotherapy concurrent with RT, which must be completed within 1 to 42 days prior to randomization and the first dose of IP.
- The chemotherapy regimen must contain platinum and etoposide, as per local standard-of-care regimens
- For patients who are recovering from toxicities associated with initial treatment or for whom PCI is indicated by local standard of care, the first dose of IP may be delayed by up to 42 days from the end of the CRT
- Radiotherapy must have commenced no later than the end of Cycle 2 of chemotherapy
- Receipt of 3 cycles of platinum-based chemotherapy concurrent with RT will be permitted if the patient achieved disease control and Investigators judge no additional benefit will be expected with additional cycle of chemotherapy
- Patients must have achieved CR, PR, or SD and not have progressed following definitive, platinum-based, concurrent CRT.
- Received a total dose of radiation of 60 to 66 Gy for standard QD radiation schedules or 45 Gy for hyperfractionated BID radiation schedules. Sites are encouraged to adhere to mean organ radiation dosing as follows:
- Mean lung dose <20 Gy and/or V20 must be <35%
- Heart V50 <25%
- Adequate organ and marrow function independent of transfusion, infusion, or growth factor support for at least 14 days prior to screening, defined as in exclusion criteria
- Must have a life expectancy of at least 12 weeks
Key exclusion criteria
- Mixed SCLC and NSCLC histology
- Extensive-stage SCLC
- Patients with Grade ≥2 pneumonitis from prior CRT
- History of allogeneic organ transplantation
- Active or prior documented autoimmune or inflammatory disorders (including inflammatory bowel disease [eg, colitis or Crohn’s disease], diverticulitis [with the exception of diverticulosis], systemic lupus erythematosus, Sarcoidosis syndrome, or Wegener syndrome [granulomatosis with polyangiitis, Graves’ disease, rheumatoid arthritis, hypophysitis, uveitis, etc]). The following are exceptions to this criterion:
- Patients with vitiligo or alopecia
- Patients with hypothyroidism (eg, following Hashimoto syndrome) stable on hormone replacement
- Any chronic skin condition that does not require systemic therapy
- Patients without active disease in the last 5 years may be included but only after consultation with the Study Physician
- Patients with celiac disease controlled by diet alone
- Uncontrolled intercurrent illness, including but not limited to, ongoing or active infection, symptomatic congestive heart failure, uncontrolled hypertension, unstable angina pectoris, uncontrolled cardiac arrhythmia, active ILD, serious chronic GI conditions associated with diarrhea, or psychiatric illness/social situations that would limit compliance with study requirements, substantially increase risk of incurring AEs or compromise the ability of the patient to give written informed consent
- History of another primary malignancy except for:
- Malignancy treated with curative intent and with no known active disease ≥5 years before the first dose of IP and of low potential risk for recurrence
- Adequately treated non-melanoma skin cancer or lentigo maligna without evidence of disease
- Adequately treated carcinoma in situ without evidence of disease
- History of leptomeningeal carcinomatosis
- History of active primary immunodeficiency
- Active infection including tuberculosis (clinical evaluation that includes clinical history, physical examination and radiographic findings, and tuberculosis testing in line with local practice), hepatitis B (known positive HBV surface antigen [HBsAg] result), hepatitis C (HCV), or human immunodeficiency virus (positive HIV 1/2 antibodies). Patients with a past or resolved HBV infection (defined as the presence of hepatitis B core antibody and absence of HBsAg) are eligible. Patients positive for HCV antibody are eligible only if polymerase chain reaction is negative for HCV RNA.
- Any unresolved toxicity NCI Common Terminology Criteria for Adverse Events (CTCAE) Grade ≥2 from previous CRT with the exception of alopecia, vitiligo, and the laboratory values defined in the inclusion criteria:
- Patients with Grade ≥2 neuropathy will be evaluated on a case-by-case basis after consultation with the Study Physician.
- Patients with irreversible toxicity not reasonably expected to be exacerbated by treatment with durvalumab or tremelimumab may be included only after consultation with the Study Physician.
- Brain metastases or spinal cord compression. All patients will have an MRI (preferred) or CT, preferably with IV contrast of the brain, prior to study entry.
- Mean QT interval corrected for heart rate using Fridericia’s formula (QTcF) ≥470 ms calculated from 3 ECGs (within 15 minutes at 5 minutes apart)
- Known allergy or hypersensitivity to any of the study drugs or any of the study drug excipients
- Patients who received sequential CRT for LD-SCLC (no overlap of RT with chemotherapy)
Contact opnemen over een studie
Neem contact op voor meer informatie over de studies van de afdeling thoracale oncologie van Amsterdam UMC.